Haplotype inference¶
Sometimes variants are linked on the same genome. Since Illumina uses short reads, if variants that are present on the same genome are too distant it is impossible to infer the linkage. But, if linked variants are proximal, it is possible to infer this linkage by looking at the fraction of reads with both variants present, compared to reads where only one variant or no variant is present. By performing statistical tests for independence on the two variants it is possible to “link” them to the same genome (at least, to a certain frequency within the population). After pairs of loci were linked, if haplotypes do not share variants, it is possible to merge several pairs together to a longer haplotype.
Warning
If the population is too heterogeneous (that is, many divergent sequences of the same genomic region are present), the haplotypes inference is expected to be inaccurate due to possible sharing of variants between different strains.
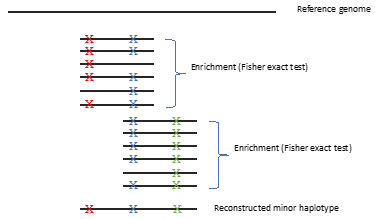
Schematic of haplotype inference script.
Finding pairs of linked loci¶
In order to find pairs of linked loci, it is required to first run the AccuNGS
Base calling stage. An intermediate of this stage is a file with the suffix
.good_reads.mutations.list. This file lists all individual mutations found
for each read. By combining this file with the BLAST results file (to obtain
the number of mutations covering each two loci), it is possible to generate a
contingency table for all pairs of variants and then test for independence,
using Fisher’s exact test. The python script variants_on_same_read.py
accepts the two input files and outputs a file with all combinations of two
sites, the counts of all variants and the results of the independence tests,
along with an associated p-value.
Required parameters¶
| Parameter name | Type | Description |
|---|---|---|
| blast_output | String | The output of the blast step of Base calling stage |
| mutation_list | String | The list file with the suffix .good_reads.mutations.list obtained during base calling step of Base calling stage |
| pos | Integer | The position x to consider all possible pairs between x and positions in the interval (x, x+250] |
| freqs_file | String | The output of Base calling stage |
Usage¶
python haplotypes/variants_on_same_read.py ${blast_output} ${mutation_list} ${pos} ${freqs_file} > pos_${pos}.txt
Output¶
The output of this step is a comma-separated file with five columns:
| Parameter name | Type | Description |
|---|---|---|
| first_pos (column 1) | Integer | The ${pos} input |
| second_pos (column 2) | Integer | The second position in the pair. Always greater than first_pos |
| odds_ratio (column 3) | Float | The prior odds ratio |
| p_value (column 4) | Float | The p-value for seeing this result if the two variants are independent |
| frequency (column 5) | Float | The frequncy of reads from all reads covering first_pos and second_pos with two variants |
Linked pairs to haplotypes¶
Once two loci were linked to the same genome (and to a given frequency), if one locus is also linked with another locus it is possible to merge the two pairs together to a longer haplotype. This can be done iteratively over all pairs to generate long haplotypes, which are a collection of variants that are presumably on the same haplotype (under the assumption that different haplotypes in the sample do not share variants).
Usage¶
The python script haplotypes/co-occurs_to_stretches.py allows merging pairs
of linked variants to longer (partial) haplotypes.
It requires the output of the linked pairs step, for all positions, as a single file.
Example usage:
python haplotypes/co-occurs_to_stretches.py linked_pairs.txt
Required parameters¶
This script function accepts four parameters:
| Parameter name | Type | Description |
|---|---|---|
| linked_pairs | String | Path to a file with all linked pairs identified using variants_on_same_reads.py |
-o / --out |
String | The output file path. Defaults to linked_pairs.stretches.out |
-p / --max_pval |
Float | The maximum p-value to consider a pair as valid |
-d / --distance |
Float | The distance between the frequencies of two pairs to consider them both on the same haplotype |
Output¶
The output is a tab-delimited file similar to this example output file.
Its columns are the following:
| Column name | Type | Description |
|---|---|---|
| Pos1 | Integer | First position in pair |
| Pos2 | Integer | Second position in pair |
| Freq | Float | The frequency that Pos1 and Pos2 variants were linked, from all reads mapped with both positions |
| Stretch | Integer | The index of this stretch, starting from 0 and above. Stretches are ordered by their meandist, descending |
| meandist | Float | The mean Freq for all pairs participating in Stretch |